Investigators at the Center for Neurosciences at The Feinstein Institutes for Medical Research have pioneered in the development and validation of disease-related covariance patterns in patients with a wide variety of neurodegenerative disorders. The way each of these characteristic patterns, or networks, is expressed in the brain of a patient can be quantified prospectively by a single number. The number, or subject score, that results serves as a robust, disease-specific biomarker.
Furthermore, it has been consistently shown that such imaging biomarkers correlate significantly with independent indices of dopaminergic function, as well as with clinical measures of disease severity. They can facilitate early differential diagnosis of parkinsonism as well as characterize unique features of neurogenetic disorders such as Huntington’s disease and dystonia. More, they provide a window for clinicians into the stages and time course of disease progression, as well as a measure for researchers of the patient’s responses to medical and neurosurgical interventions.
The multitracer methodology developed by CFN has led to critical insights on the interactions between localized dopaminergic deficits and the impairments in related brain networks underlying motor and cognitive dysfunction in neurodegenerative disorders.
The research summaries given here are grouped as follows:
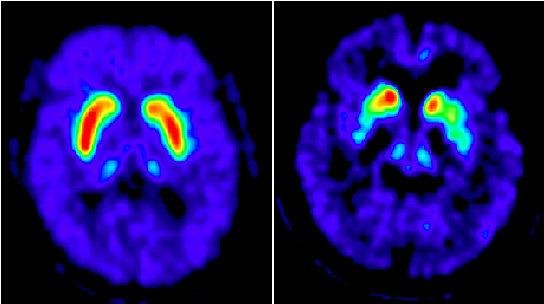
PET scans highlight the loss of dopamine storage capacity in Parkinson’s disease. In the scan of a disease-free brain, made with [18F]-FDOPA PET (left image), the red and yellow areas show the dopamine concentration in a normal putamen, a part of the mid-brain. Compared with that scan, a similar scan of a Parkinson’s patient (right image) shows a marked dopamine deficiency in the putamen.
Parkinson’s disease is one of the most common neurodegenerative diseases, a chronic, progressive movement disorder that affects the lives of at least half a million Americans. The initial symptoms are subtle—fatigue, muscular cramping, stiff arms and legs—beginning, on average, in the patient’s mid-50s. Difficulty with the sense of smell also occurs in as many as 90 percent of Parkinson’s patients, and it may predate clinical Parkinsonism by at least four years.
As the disease progresses, purposeful movement slows (bradykinesia), stiffness and tremors often develop, and balance becomes more difficult. These symptoms often first appear on only one side of the body, and the initial asymmetry worsens with time. Other motor symptoms include stooped posture, a shuffling gait, difficulty with speech, and small handwriting (micrographia).
People with PD also experience significant non-motor symptoms, including mood changes such as depression, anxiety, and apathy, impulsive or compulsive behavior, cognitive impairment, sleep disturbances, and autonomic dysfunction.
The disease is caused by the degeneration of neurons in the substantia nigra of the mid-brain that produce the neurotransmitter dopamine. The loss of dopamine leads to changes in the basal ganglia circuitry, and ultimately to abnormal cortical activation patterns.
The dopamine-precursor levodopa is initially quite effective in treating the major motor symptoms of PD. Eventually, however, its potency wears off, and a disruptive motor side-effect arises, known as levodopa-induced dyskinesias (LID).
Parkinson’s disease patients, their families, friends, and caregivers will find links to more information about living with the disease here.
For a list of references to recently published research papers on Parkinson’s disease, please click here.
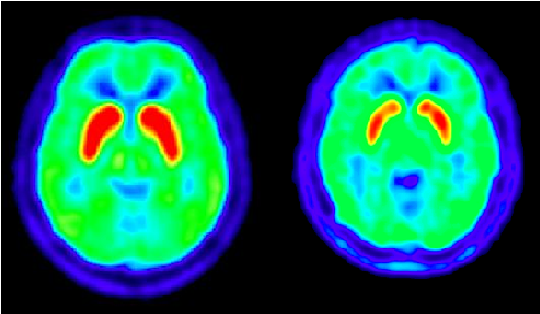
PET scan of a healthy person’s brain (left image), compared with a PET scan of a patient with Huntington’s disease, 3.6 years into the progression of the disease (right image), shows the loss of dopamine D2 receptors in the mid-brain. The scans were made with [11-C]-RAC PET.
Huntington’s disease (HD) is a hereditary neurodegenerative disorder that afflicts most of its sufferers early in adult life, causing progressive deterioration in motor, cognitive, and behavioral functioning. Mounting clinical, imaging, and pathological evidence suggests that the neurodegenerative process underlying HD begins many years before it manifests the signs and symptoms needed for a confirmed clinical diagnosis. Yet an intervention during those preclinical years would have far greater impact than any later therapy in slowing or halting the neurodegeneration.
Although no interventions are yet available to alter the course of HD, much work is under way to develop them. Numerous clinical trials are ongoing to evaluate the potential neuroprotective effects of experimental therapies in patients with manifest HD. A natural follow-up would be to study the effects of these agents on presymptomatic carriers of the mutated HD gene: those with preclinical Huntington’s disease (pHD). Nevertheless, undertaking clinical trials in a group of clinically normal individuals can be problematic.
One major difficulty is defining the best way to measure trial outcomes. HD outcomes are currently measured with such tools as the Unified Huntington’s Disease Rating Scale (UHDRS), but clinical measures of disease symptoms are obviously not useful for assessing clinically unaffected individuals. Another possible measure is phenoconversion—the time it takes a patient to progress from pHD to diagnosed HD—but that, too, may be impractical: some pHD subjects in clinical trials may be many years from developing unequivocal signs of HD. For those reasons there has been a concerted effort to identify reliable biomarkers for measuring disease progression in pHD subjects.
The Center for Neurosciences at The Feinstein Institute for Medical Research is utilizing a new network modeling strategy designed for the analysis of longitudinal brain-imaging data. Center investigators have identified and validated an HD-related progression pattern (HDPP) in resting state metabolic scans of presymptomatic (or “premanifesting”) carriers of the HD gene mutation. Preliminary data suggest that when an HDPP is detected and changes with time, it reflects functional changes occurring across the whole brain. Thus the HDPP is likely to be more sensitive to disease progression than regional imaging biomarkers.
For a list of references to recently published research papers on Huntington’s disease, please click here.
Primary dystonia, which often begins in late childhood or adolescence, has traditionally been attributed to dysfunction of the basal ganglia, but no specific histopathological lesions of these structures are evident in patients with the disease. In fact, the challenge of primary dystonia is reflected in its clinical definition: the presence of involuntary, sustained muscle contractions in the absence of identifiable brain lesions. This lack of clear neuropathology has made dystonia difficult to understand and or to treat.
Recent work at the Center for Neurosciences supports a new paradigm for understanding dystonia: as a neurodevelopmental circuit disorder. Using new magnetic resonance diffusion tensor imaging (DTI), Center investigators examined motor circuit activity in patients with manifest dystonia, as well as in nonmanifesting carriers of the dystonia gene mutation DYT1. Surprisingly, the investigators discovered that both groups exhibited disruptions of the cerebellar motor pathway.
Among patients with manifesting dystonia, the investigators found one lesion that seemed to disrupt the flow of neural signals from one part of the motor pathway to the other.
Among the nonmanifesting gene carriers, however, the investigators found a lesion in a second place along the motor pathway, downstream from the first, in the part of the pathway connecting directly to the motor cortex.
The second lesion ought to have been just as effective as the first in disrupting neural signals. Yet the correct signals were getting through. Why?
Further studies have shown that this second disruption of the pathway actually canceled out the effects of the first lesion, by completely blocking the transmission of the aberrant cerebellar output of the first lesion.
With no disruptive neural noise reaching the motor cortex, only clear signals from other, undisrupted neural pathways could get through.
This work has given rise to a new model for the dysfunction in motor circuitry that underlies dystonia.
Center investigators are measuring serial changes in brain network activity as the treatment response develops; identifying patterns of microstructural change at baseline that correlate with clinical treatment response; and developing quantitative, preoperative imaging descriptors to predict the responses of individual patients to surgical treatment.
Specifically, Center investigators are characterizing motor circuit abnormalities in hereditary primary dystonia by examining structure-function relationships in manifesting and non-manifesting carriers of mutated DYT1, as well as the DYT6 mutation. Both pathway microstructure and brain circuit function are assessed with positron emission tomography (PET) and functional MRI (fMRI). PET scanning maps a patient’s blood flow in the brain during task performance as well as in the rest state. Functional MRI enables investigators to localize task-related neural activation responses.
Circuit abnormalities in sporadic dystonia are identified via diffusion tensor imaging/ tractography MRI studies and brain activation experiments. The results are compared to those from patients with hereditary forms of the disease.
Investigators also want to understand how deep brain stimulation (DBS) works in dystonia— and to identify reliable predictors of a patient’s response to the therapy. DBS can be effective in treating severe primary dystonia, but not all patients benefit equally, and symptoms can re-emerge after an initial period of abatement.
For a list of references to recently published research papers on dystonia, please click here.
Identifying and making clinical use of disease-specific networks in the brain depend on imaging both functional and anatomical substrates. Functional imaging relies on positron emission tomography (PET) and functional magnetic resonance imaging (fMRI); anatomical imaging is done with structural magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI).
PET imaging is based on the use of multiple radiotracers for the measurements of impaired neurochemical (e.g., dopamine or serotonin) function and altered cerebral metabolism and blood flow. More recently, multitracer PET imaging has also made it possible to visualize protein aggregates such as amyloid depositions that are associated with dementia.
PET imaging procedures with novel molecular radioligands have been widely used for identifying local changes in presynaptic and postsynaptic dopaminergic function in neurodegenerative diseases. As a common presynaptic marker of dopamine storage capacity in the nigrostriatal projection pathway, the radiotracer [18F]-FDOPA was the first to demonstrate dopamine deficiency in the putamen in PD patients. At early stages of the disease course, this deficiency is evident in the posterior putamen contralateral not only to the clinically affected limbs but also to the clinically unaffected limbs.
The radiotracer [18F]-FPCIT offers another means for detecting the loss of dopamine nerve terminals in PD patients: it serves as a presynaptic marker (in fact, one of many) of dopamine transporter (DAT).
It has been reported that FDOPA uptake and DAT binding in the striatum are often interchangeable as neurobiological descriptors of dopaminergic dysfunction in parkinsonism, based on in vivo and in vitro investigations in humans as well as in primate and small animal models of PD. These measures correlate with the severity of clinical symptoms in individual patients and are modulated by disease-modifying therapeutics.
Local changes in postsynaptic dopaminergic function have also been detected in patients with HD and dystonia. Dopamine D2 receptors, for instance, can be imaged via PET with the radiotracer [11C]-raclopride (RAC), one of many D2 markers. In carriers of the mutated HD gene, the loss of striatal dopamine is evident in RAC-PET scans even long before the onset of clinical symptoms. The amount of loss, moreover, is related to the expected number of years before onset.
Similar losses of striatal D2 receptors occur in carriers of the DYT1 and DYT6 genes, both implicated in dystonia. Those losses are more severe in the DYT6 carriers than in the DYT1 carriers, just as patients with clinical symptoms of dystonia exhibit more severe losses than presymptomatic carriers do, regardless of genotype.
PET imaging has also been used for mapping changes in regional cerebral glucose metabolism and blood flow with [18F]-FDG ([18F] fluorodeoxyglucose) and [15O]H2O ([15O] water), respectively. These two techniques are most useful for highlighting disease-specific brain networks and their modulation–either by the disease process or by experimental therapeutics.
Two other PET radiotracers, [11C]-PIB and [18F]-FDDNP, can be used to quantify brain amyloid accumulation in patients with mild cognitive impairment as well as with dementia.
All the PET radiotracers described above are produced in the cyclotron/radiochemistry laboratory at CFN and routinely imaged in patients’ brains via PET. Those images are highly valuable for measuring progressive changes in brain function, which are in turn important for assessing the effectiveness of neuroprotective and symptomatic agents in clinical trials.
For a list of references to recently published research papers on brain imaging in neurodegenerative disorders, please click here.